Our Focus
We take S protein as the research focus and take coronavirus recombination as the exploration direction.
| Coronavirus Resources | Metagenomics Resources |
| ======= software ======= |
===== Data ===== |
======= Papers ======= |
====== Video ====== |
======== Join us ======== |
============= Popular science ============= |
========== Our advance ========== |
=========== Member Login =========== |
========== |
 |
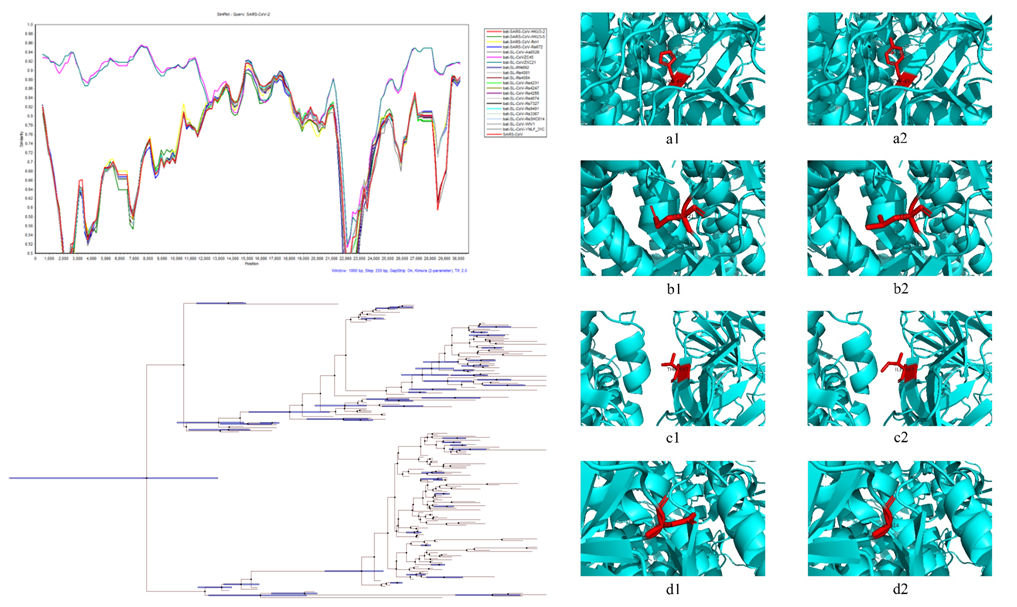
Molecular Evolution and Variation Characteristics of SARS-CoV-2 Spike Protein
Abstacts:SARS-CoV-2 is a new type of β-coronavirus newly discovered at the end of 2019, which is highly pathogenic and
poses a serious threat to human health. In this paper, through the
bioinformatics analysis of the new virus, the molecular evolution and
mutation of its spike protein are studied. Using 213 SARS-CoV-2 samples
obtained from the United States, the evolution rate of the whole
genome sequence and spike protein sequence was analyzed by MCMC method,
and it was found that the evolution rate of the whole genome was higher
than that of the spike protein region. Compared with previous studies,
the results obtained are lower in evolution rate. Because it is highly
similar to the sequences of various bat SARS-like coronaviruses and
SARS-CoV in GenBank, we established their phylogenetic tree to study
the selective evolution of SARS-CoV-2 spike protein. According to the
research results, there is a clear positive selection in the spike
protein of SARS-CoV-2, and 12 statistically significant positive
selection sites were found, of which 3 are located in the receptor
binding domain of the spike protein. In addition, sequence alignment
results show that multiple non-synonymous mutation sites have appeared
in 213 SARS-CoV-2 samples. According to sequence homology and amino
acid physical properties, there are mutations that can cause the
structure and function of spike protein.
Bioinformatic analysis of key pathogenic sites of human coronavirus NL63
Abstract:By comparing themultiple sequence alignment results of both complete genome sequence and receptor-binding Spike (S) protein of human coronavirus NL63, we discovered that within the genome sequence the similarity of S protein was low, thus meaning there were mutations existing within S protein region. By contrast, there were only three amino acid mutations in the amino acid sequence of S protein. Therefore, in this dissertation, we further characterized the S protein of HCoV-NL63 and located key residues of RBD-receptor association through the virus S protein phylogeny analysis, using Pymol to construct protein conformation on three mutant sites, analyzing its effects on HCOV-NL63 S protein and using ProSNEx to determine hot spot residues in the viral-receptor binding domain. Our results showed that: the S protein of HCoV-NL63 was highly-conserved and the mutations had occurred within the S protein played an important role in stabilizing the structure of S protein. As HCoV-NL63 showed that the more stable the S protein was, the more affinitive it becameto bind human receptor ACE2. Therefore, we believe that HCOV-NL63 will coexist with human for a long time and its invasion and infection ability will gradually increase. The Aa353 and Aa354 are the main binding residues on receptor ACE2and Aa494-499 and Aa535-540 within RBD were important for receptor binding and virus entry.
3. Jingying Zhao, Shanghai Ocean University, Biosciences, 2016 undergraduate
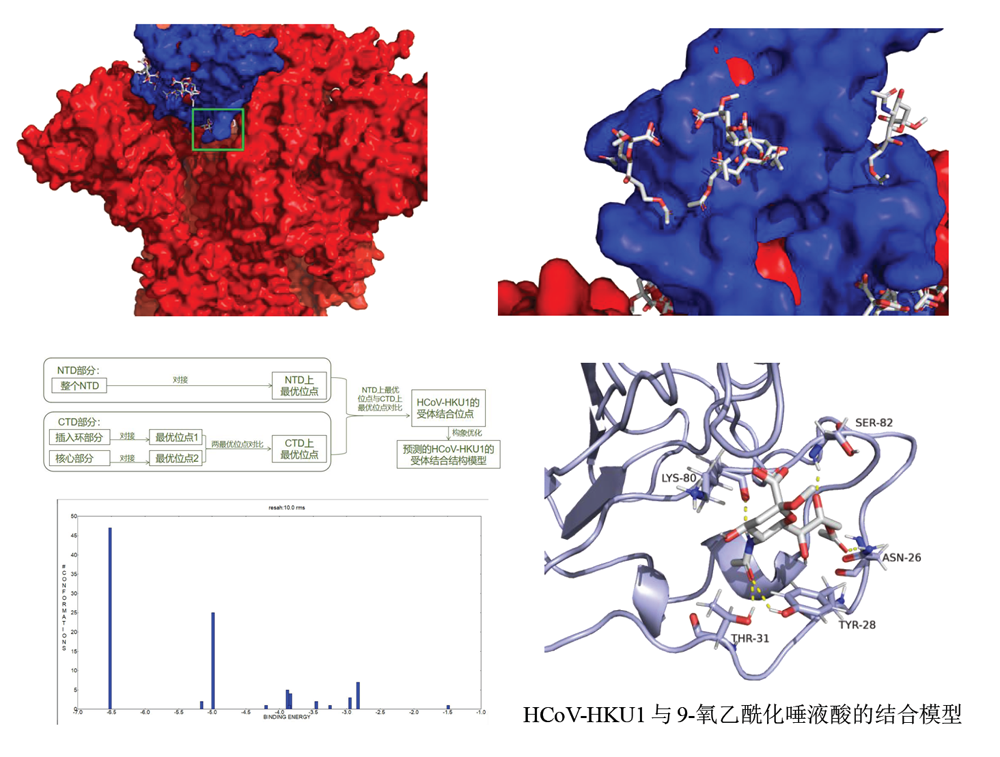
Prediction of HCoV-HKU1 receptor binding site based
on molecular docking technology
Abstacts:In this paper, molecular docking was performed by Autodock software, and receptor binding site of HCoV-HKU1 was found at a position B on the NTD of its S protein S1 subunit ( corresponding to amino acid residues 80-95, 25-32 ). And obtained the binding model of site B with 9-O-acetylated sialic. This article also discusses the ability of the CTD of the S1 subunit of the HCoV-HKU1 S protein to bind to 9-O-acetylated sialic. It is believed that the purified CTD does have the ability to bind to 9-O-acetylated sialic, but in the intact S protein, its CTD exposed part is not as good as NTD in binding to 9-O-acetylated sialic.
4. Yin Zheng, Haiyang Ocean University, Marine Biology, 2016 undergraduate
Bioinformatics analysis of bat coronavirus genetic recombination events
Abstract:In order to more comprehensively explore the genetic recombination events occurring between bat coronaviruses and to further understand and explore the animal origin of human coronaviruses, this study used the phylogenetic tree construction function and RDP4 of MEGA-X software in the bioinformatics software package The 7 recombination detection algorithms included in the software phylogenetically developed the entire genome sequence of 19 coronavirus strains with human, bat, pangolin, and civet as the host between 2003 and 2018 Tree construction and genetic recombination testing. The results of gene recombination analysis detected a total of 2 recombinant strains and a total of two recombination events. The two recombinant strains were MG772934 and MG772933, both of which were bat host coronavirus sequences collected in Zhoushan. The recombination area involved The edited fragments of the genome sequence are non-structural proteins, and the reorganization has actually changed the branch of the evolutionary tree where the recombinant strain is located; the parent of a recombination event is the presence of a coronavirus of the same species host in the non-recombinant strain, indicating that the virus may be in the boast In the process of transmission, there are repeated infections or sharing of receptors, but the specific behavior still needs further study. The above results indicate that genetic recombination may change the ability of coronavirus to spread across species and affect the genetic evolution of coronavirus. This shows that the genetic recombination between coronaviruses can promote the emergence of new coronavirus strains.

We take S protein as the research focus and take coronavirus recombination as the exploration direction.